Períodos transitorios Anexo XVI MDR. ¡No te quedes atrás!
Los períodos transitorios Anexo XVI del Reglamento (UE) 745/2017 (MDR) siguen siendo la principal fuente de quebraderos de cabeza de los operadores económicos (fabricantes, importadores, distribuidores…) que comercializan productos “sin finalidad médica prevista”, pero regulados ahora por el MDR
En este artículo vamos a aclarar las fechas de estos periodos transitorios de los Productos Sanitarios sin finalidad médica recogidos en el Anexo XVI del reglamento (UE) 2017/745.
Antes de entrar en materia y ver a qué productos afecta y de qué manera, es muy importante tener en cuenta que la Comisión, mediante el Reglamento de Ejecución (UE) 2023/1194 (20 de junio de 2023), modificó el Reglamento de Ejecución (UE) 2022/2346 para ajustar las disposiciones transitorias aplicables por lo que es de vital importancia tener el calendario actualizado para no equivocarnos.
En primer lugar, revisaremos, de manera general, cuáles son esos productos incluidos en el Anexo XVI.
¿Qué productos incluye el Anexo XVI y por qué tienen “especificaciones comunes”?
El MDR incluye en su Anexo XVI determinados grupos de productos sin finalidad médica prevista, pero que pueden implicar riesgos para la salud comparables y, por ello, se regulan de forma similar. El Reglamento de Ejecución (UE) 2022/2346 establece especificaciones comunes (Common Specifications, CS) para estos grupos, incluyendo (entre otros):
- Lentes de contacto.
- Productos destinados a ser introducidos total o parcialmente en el cuerpo por medios invasivos quirúrgicos para modificar la anatomía (con exclusiones indicadas en el texto).
- Rellenos faciales u otros rellenos dérmicos o de mucosas mediante inyección (u otros medios de introducción).
- Equipos para reducir/retirar/destruir tejido adiposo (p. ej., liposucción, lipólisis, lipoplastia).
- Equipos que emiten radiación electromagnética de alta intensidad (láser, IPL, etc.) para usos estéticos como rejuvenecimiento, depilación o eliminación de tatuajes.
- Equipos para estimulación cerebral mediante corrientes o campos magnéticos/electromagnéticos que penetran en el cráneo para modificar la actividad neuronal.
Estas especificaciones comunes abarcan elementos esenciales de gestión de riesgos y requisitos del Anexo I del MDR para asegurar seguridad y funcionamiento.
Ahora bien, una vez tenemos claro que nuestro producto entra dentro de este anexo, vamos a ver cómo podemos mantener el producto en el mercado teniendo en cuenta las diferentes casuísticas existentes.
La clave del régimen transitorio: ¿puedo seguir introduciendo el producto en el mercado?
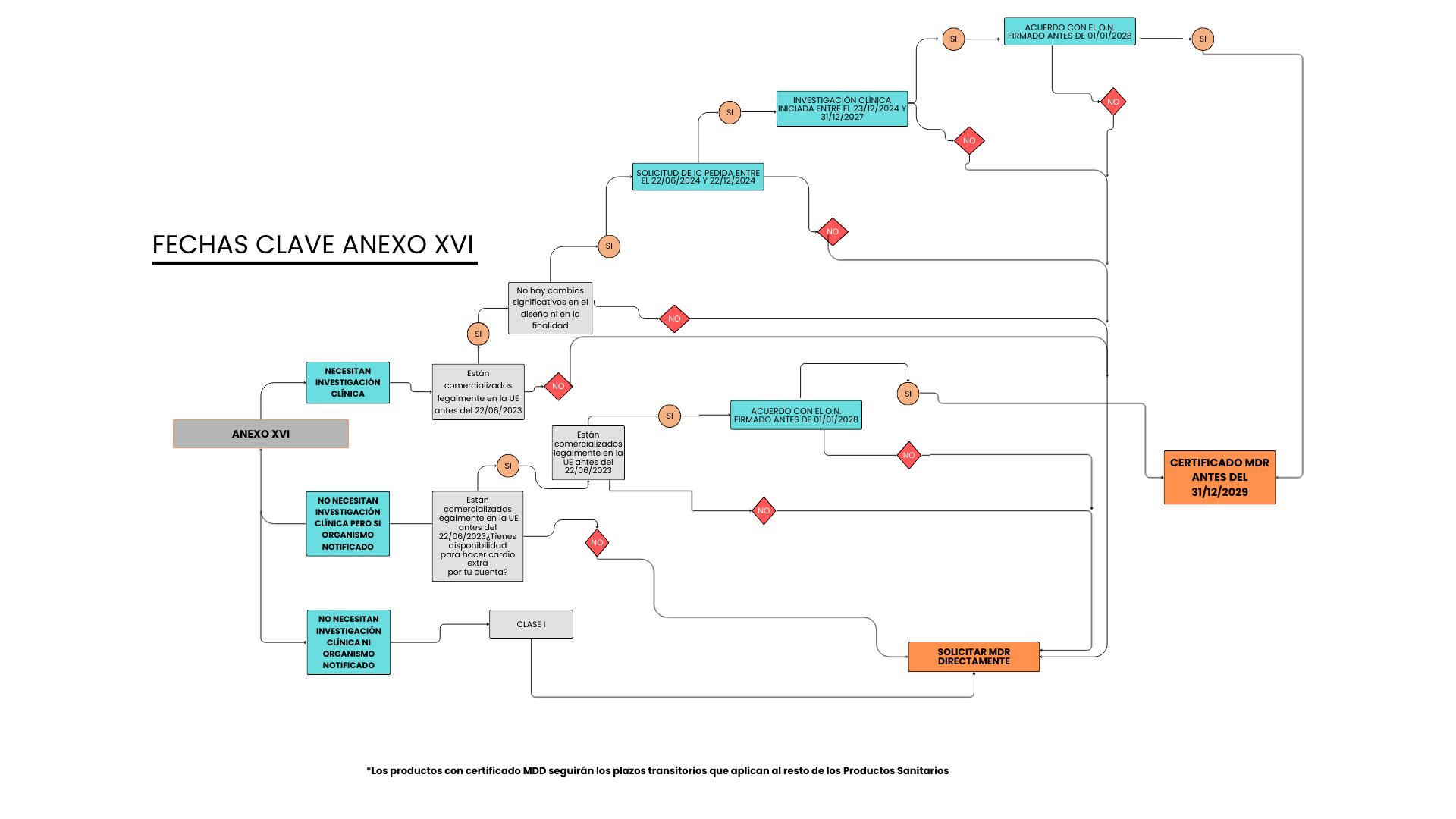
Para beneficiarte de los períodos transitorios Anexo XVI MDR, el Reglamento establece una regla base muy clara: solo pueden acogerse los productos que:
- Ya estuvieran comercializados legalmente en la UE antes del 22 de junio de 2023, y
- Sigan cumpliendo la normativa UE/nacional que les aplicaba antes de esa fecha, y
- No hayan sufrido cambios notables en el diseño ni en la finalidad prevista.
A partir de ahí, el calendario cambia según el escenario regulatorio en el que se encuentre tu producto.
Escenario 1: productos que necesitan investigación clínica para generar datos clínicos
Si el fabricante está investigando clínicamente el producto o prevé realizar investigaciones clínicas para sustentar la evaluación clínica (y además la evaluación de la conformidad requiere organismo notificado), entonces el producto puede introducirse en el mercado o ponerse en servicio hasta el 31 de diciembre de 2029, siempre que cumpla las condiciones base anteriores.
Pero atención: el Reglamento “endurece” progresivamente las condiciones a medida que avanza el tiempo:
- Del 22/06/2024 al 22/12/2024: debes haber recibido una notificación del Estado miembro conforme al art. 70(1) o 70(3) MDR confirmando que la solicitud de investigación clínica está completa y entra en el ámbito del MDR.
- Del 23/12/2024 al 31/12/2027: se debe haber iniciado la investigación clínica.
- El 01/01/2028 a más tardar, el fabricante y el organismo notificado han firmado un acuerdo escrito para realizar la evaluación de la conformidad (referencia a Anexo VII, sección 4.3).
Este es el punto donde muchas empresas fallan: no basta con “tener intención”; en 2028–2029 debes poder demostrar una relación contractual activa con organismo notificado para la evaluación. Si no, por mucho que hayas cumplido con los anteriores plazos, tu producto no podrá ser fabricado ni importados.
Escenario 2: productos que no necesitan investigación clínica, pero con organismo notificado obligatorio
Si el fabricante no prevé realizar investigaciones clínicas, pero el producto requiere participación de organismo notificado conforme al art. 52 MDR, entonces puede introducirse en el mercado o ponerse en servicio hasta el 31 de diciembre de 2028, cumpliendo las condiciones base.
Además:
- El 01/01/2027 a más tardar,el fabricante y el organismo notificado han firmado un acuerdo escrito para la evaluación de la conformidad.
En la práctica, esto significa que 2026 es el año “bisagra” para no llegar tarde: selección de organismo notificado, planificación de expediente y cierre de contrato.
¿Qué se considera “cambio significativo” y por qué importa tanto?
La expresión es crítica porque si cambias el diseño o la finalidad prevista de forma significativa, pierdes el derecho al periodo transitorio.
Como referencia práctica, la guía MDCG 2020-3 Rev.1 explica el concepto de “cambios significativos en diseño o finalidad prevista” en el marco del art. 120 MDR y cómo evaluar caso por caso (enfoque útil para evitar interpretaciones laxas).
Recomendación operativa: documenta un procedimiento interno de “Evaluación de cambios” (impacto en seguridad, desempeño, reclamaciones, materiales, software, energía emitida, IFU, etc.) antes de modificar el producto durante el período transitorio.
Checklist de preparación (recomendado) para no perder los plazos:
Para gestionar bien los períodos transitorios Anexo XVI MDR, estas acciones suelen ser las más efectivas:
- Confirmar evidencia de comercialización legal en UE antes del 22/06/2023 y mantenimiento de cumplimiento previo.
- Clasificación y ruta de evaluación: determinar si requiere ON y si habrá investigación clínica (o no).
- Plan regulatorio con hitos: incorporar las fechas 2026, 2027, 2028, 2029 en un cronograma realista.
- Estrategia de evidencia clínica (si aplica): plan de investigación clínica, plazos, inicio y seguimiento.
- Organismo notificado: preselección, capacidad, tiempos de contrato y auditoría; objetivo: acuerdo escrito antes de 2027 o 2028 según escenario.
- Control de cambios: blindar el periodo transitorio evitando cambios notables sin evaluación formal.
Gestionar correctamente los períodos transitorios Anexo XVI MDR no es “apurar plazos”, sino de planificar evidencia, expediente y organismo notificado para llegar a tiempo con un camino auditado y defendible.
Desde AMConsulting podemos ayudarte te encuentres en la fase que te encuentres, no desaproveches que aun queda tiempo y contáctanos.